Next: MCMC based results Up: What is the probability Previous: Introduction
The causal model used in this analysis
is implemented in the Bayesian network
of Fig. ![[*]](crossref.png) .
.
.
Then, there is the question of how to relate the numbers
of infectees to the numbers of the participants in the trial.
This depends in fact on several variables, like the
prevalence of the virus in the population(s) of the involved people,
their social behavior, personal
life-style, age, health state and so on. And, hopefully,
it depends on the fact
that a person has been vaccinated or not.
Lacking detailed information, we simplify
the model introducing an assault probability ![]() , that is a
catch-all term embedding the many real life variables, apart
being vaccinated or not.
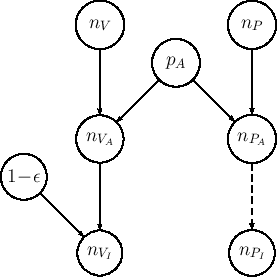
Nodes
, that is a
catch-all term embedding the many real life variables, apart
being vaccinated or not.
Nodes ![]() and
and ![]() in the network of Fig.
represent then the number of `assaulted individuals'
in each group, and they are modeled according to
binomial distributions, that is
in the network of Fig.
represent then the number of `assaulted individuals'
in each group, and they are modeled according to
binomial distributions, that is
The `assaulted individuals' of the control group
are then assumed to be all infected, and hence the
deterministic link with dashed arrow relating node ![]() to node
to node ![]() follows (indeed the two numbers are exactly
the same in our model, and we make this distinction
only for graphical symmetry with respect to the vaccine group).
follows (indeed the two numbers are exactly
the same in our model, and we make this distinction
only for graphical symmetry with respect to the vaccine group).
Instead, the `assaulted individuals' of the other group
are `shielded' by the vaccine with probability ![]() ,
that we therefore identify with efficacy,
although we shall come back at the due point about what
should be reported as `efficacy'.
The probability of becoming
infected if assaulted is therefore equal to
,
that we therefore identify with efficacy,
although we shall come back at the due point about what
should be reported as `efficacy'.
The probability of becoming
infected if assaulted is therefore equal to
![]() ,
so that node
,
so that node ![]() is related to node
is related to node ![]() by
by
The nice thing using such a tool is that we have to take care only to describe the model, with instructions whose meaning is quite transparent:5
model {
nP.I ~ dbin(pA, nP) # 1.
nV.A ~ dbin(pA, nV) # 2.
pA ~ dbeta(1,1) # 3.
nV.I ~ dbin(ffe, nV.A) # 4. [ ffe = 1 - eff ]
ffe ~ dbeta(1,1) # 5.
eff <- 1 - ffe # 6.
}
We easily recognize in lines 1. and 2. of the R code
the above Eqs. () and
(), while line 4. stands for
Eq. (). Line 6. is simply the transformation
of `
for details) with
parameters
|
MCMC results
Published results
|