Research Activities
My research interest is Time-Resolved Vibrational Spectroscopy, applied to access ultrafast structural and dynamical properties at the molecular and condensed matter levels.
While the foundations of the atomic structure theory go back to more than a century
ago, only in the recent decades the development of novel microscopy techniques have
succeeded in acquiring “photographs” of molecular compounds, with atomic structural
resolution. “Animating” these static pictures to monitor the atomic motions during the
primary processes that govern physical, chemical and biological phenomena, carrying out
a real molecular movie, is nowadays one of the greatest scientific challenges. Recording
transient atomic motions in action would allow to unveil, for example, the mechanisms ruling
phase transition processes in physics and to disclose bond breaking and recombination
events in biochemistry. At the microscopic level, since
the speed of atomic motion is ≈ 1 km s-1, the capability of measuring structural changes
of reacting species over the atomic scale (a few ångström) on timescales short enough to resolve the
reaction pathways requires ≈ 100 fs “exposure times”.
The impressive technological improvements in the last years, especially the advent of
bright femtosecond laser sources, paved the way to a direct exploration of these temporal
realms and have been foundational to ultrafast spectroscopy, in which light pulses are used
to first excite (Pump) and subsequently interrogate (Probe) a system.
Critically, the natural hindrance in this field revolves around the simultaneous need of two
key ingredients: high temporal and spectral resolutions, which are mutually compromised
due to the Heisenberg principle. Ultrafast electronic spectroscopies offer femtosecond time
resolution, but cannot provide detailed information on the geometrical configuration of
the system, due to the lack of structural sensitivity. On the other hand, vibrational spectroscopies,
used to study molecular and solid state compounds, provide excellent spatial
resolution but are ineffective on subpicosecond timescales.
In this respect, my work focuses on the development of experimental, conceptual and numerical protocls able to circumvent restrictions dictated by the Heisenberg principle for unravelling matter properties on picosecond and sub-picosecond time regimes.
The main idea is to exploit Coherent Raman Spectroscopy for combining the structural sensitivity of vibrational specroscopy with the temporal resolution that can be achieved in Non-Linear Spectroscopy investigating a system of interest with multiple light pulses.
Highlights
New insights into Coherent Raman thermometry.
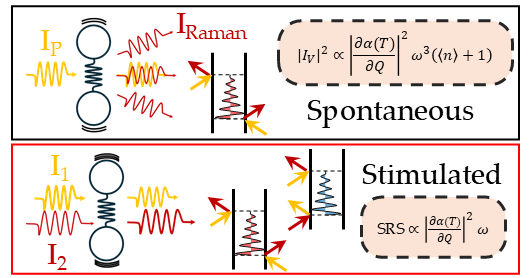
Among its diverse applications,
Raman spectroscopy is a well-established tool for contactless thermometry, enabling non-destructive measurements in molecules and solids.
Traditionally, the temperature is determined by evaluating the ratio of Stokes and anti-Stokes lines (arising from inelastic scattering processes that involve energy transfer from the electromagnetic radiation
to the matter and vice-versa) and the cross-section thermal
dependence is described using Bose-Einstein statistics.
Recently, we reported striking deviations from this behavior,
and highlighted a fundamental difference between spontaneous and stimulated (coherent) responses.
In contrast with spontaneous spectroscopy, coherent Raman exploits multiple light pulses to stimulate and probe vibrational excitations.
This approach offers several advantages, such as fluorescence-free spectra, enhanced cross sections and improved spectro-temporal resolutions.
However, it also introduces additional energy transfer channels between light and matter,
which ultimately factor out the Bose dependence.
This directly reveals a striking temperature-dependence of the molecular polarizability -
the fundamental material property underlying the Raman response -
and explains the unexpected deviations from the classical Bose model.
Mapping the polarizability's temperature dependence via stimulated Raman spectroscopy hence
represents a powerful tool to investigate vibrational energy redistribution processes,
unlocking insights that remain elusive with conventional methods.
Femtosecond Stimulated Raman Spectroscopy (FSRS) for Dummies.

Pump-probe nonlinear Raman spectroscopy represents a powerful tool to explore ultrafast physical and chemical reactions,
able to combine structural sensitivity and high temporal resolution.
We recently published a review on Nature Reviews Methods Primers dedicated to this topic, including its applicability and the innovations driving the method forward.
The work discusses the breakthroughs brought about by non-linear optical regime,
including critical considerations on the ultimate temporal resolution and capabilities to track ultrafast molecular events.
Specifically, the Primer explores whether, why, when and how
the temporal precision and frequency resolution of traditional time-resolved spontaneous Raman spectroscopy
can be surpassed by its coherent counterpart (FSRS), while still adhering to the uncertainty principle.
The fundamental concepts behind FSRS and its most common experimental approaches are presented,
critically discussing instrumentation details.
Recent applications of FSRS from physics, chemistry and biology are showcased,
demonstrating how this approach can facilitate cross-disciplinary studies.
The work serves as a practical guide for setup design and implementation, data collection, AI-based analysis and modelling.
Signs and magnitudes of excited state displacements revealed by impulsive Raman spectroscopy.
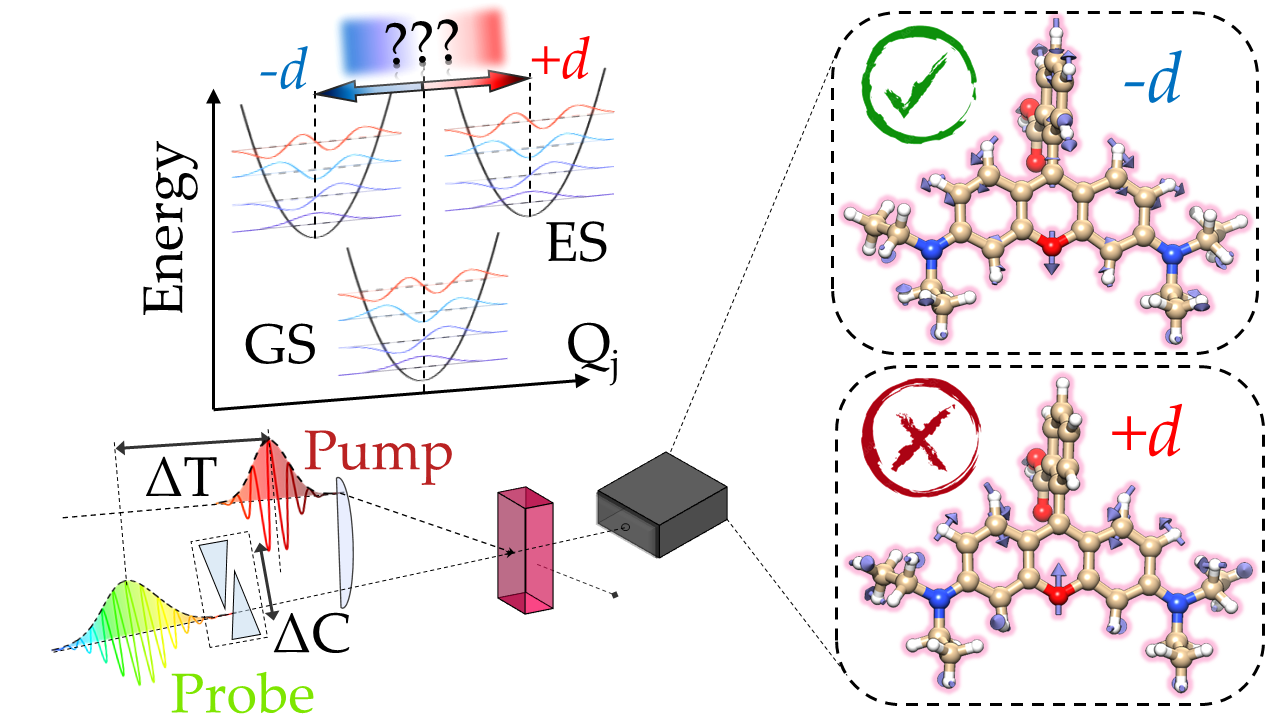
Physical and chemical reactions driven by light absorption are determined by multidimensional excited-state potential energy surfaces (PESs).
Nature has shaped excited-state PESs displaced with respect to the ground-state along specific nuclear reaction coordinates to drive the system photochemistry,
inducing specific structural rearrangements which define the biological function by positive vs negative bond length modifications and torsional re-orientations, up to formation or rupture of chemical bonds.
Such displacements are encoded in the Franck-Condon overlap integrals, which in turn determine
the resonant Raman response. Conventional spectroscopic approaches only probe their square moduli, and hence cannot
access the sign of ES displacements.
By introducing an experimental scheme, we have shown how to determine the most elusive aspect of the excited-state molecular displacements,
namely its sign relative to the ground-state.
The key to achieve this task is in the signal linear dependence on the Frank-Condon overlaps, brought about by non-degenerate resonant probe and off-resonant pump pulses,
which ultimately enables time-domain sensitivity to the phase of the stimulated vibrational coherences.
Ultrafast Energy Transfer in transition metal dichalcogenide-graphene hetrerostructures revealed by pump-probe Raman spectroscopy
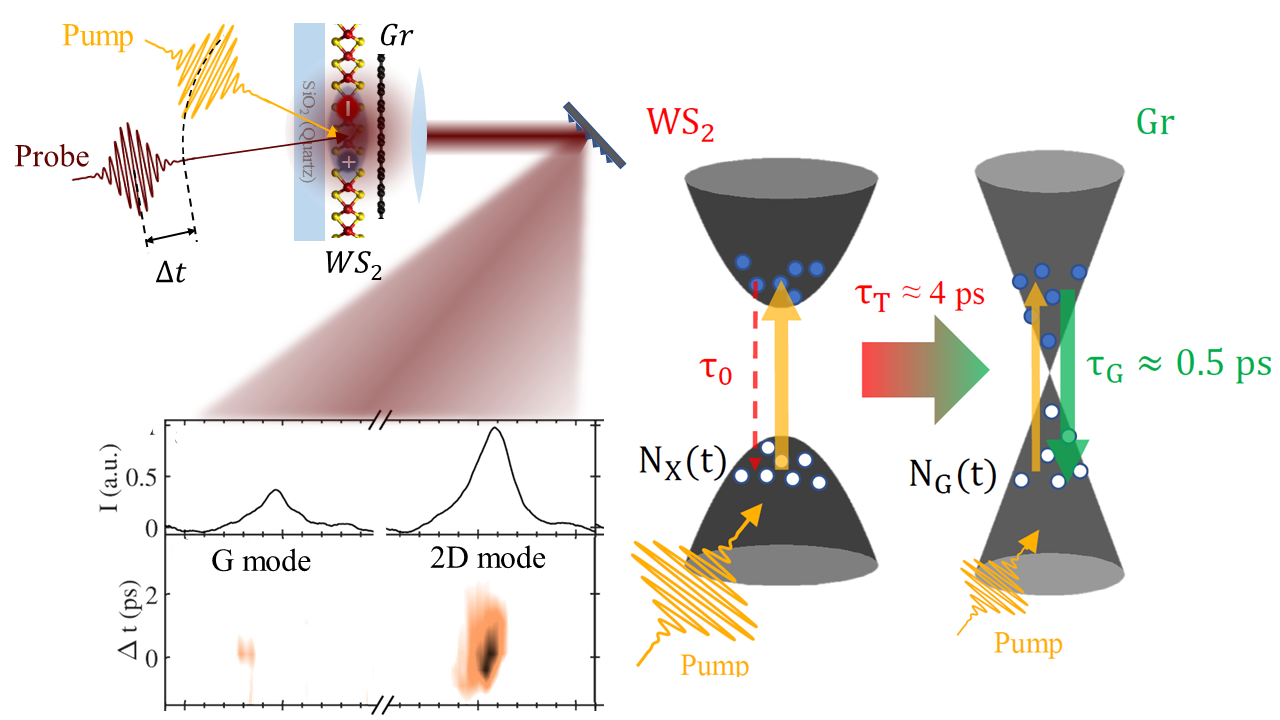
Intense light-matter interactions and unique structural and electrical properties make van der Waals heterostructures composed by graphene (Gr) and monolayer transition metal dichalcogenides (TMD) promising building blocks for tunneling transistors and flexible electronics,
as well as optoelectronic devices, including photodetectors, photovoltaics, and quantum light emitting devices (QLEDs).
An efficient energy harvesting of photoexcited hot carriers is critical for the performances of such devices.
In this respect, the way initially photogenerated excitons in the TMD are converted into an electric current in Gr is a highly controversial issue.
Exploiting a picosecond pump pulse, resonant with the WS2 monolayer absorption, we injected photo-carriers in the TMD and then monitored the graphene response with a temporally delayed probe pulse.
By tracking the picosecond dynamics of the G and 2D Raman bands, we determined the electronic temperature profile of Gr in response to TMD photoexcitation,
unveiling that the fundamental mechanism boosting the carrier injection in Gr from the TMD monolayer is an energy transfer occurring on the picosecond scale.
This indicates the existence of an additional conversion mechanism bridging such ultrafast energy transfer with the much slower charge transport involved in optoelectronics applications.
Revealing excited potential energy surfaces by Raman excitation profiles measured via time-domain Raman spectroscopy.
Spontaneous Raman spectra depend on the displacement along the normal coordinates between ground and excited potential energy surfaces (PESs),
and hence the relative intensities of the measured Raman bands encode information on the PESs relative displacement.
Critically, in order to extract such molecular information, several spectra have to be recorded scanning the Raman pump wavelength across the absorption profile,
with the detection of the experimental signals that is typically hampered by the overwhelming fluorescent background.
Most importantly, spontaneous Raman spectroscopy cannot be applied to monitor ultrafast chemical reactions on electronically excited states.
Recenlty we have shown how to circumvent these limitations introducing an approach based on time-domain impulsive Raman scattering:
a femtosecond pulse impulsively launches nuclear wave packet motions in the system under investigation and then their couplings with an arbitrary excited state potential
is measured by a resonant Raman process enabled by a delayed femtosecond probe pulse.
A perturbative treatment of the scattering process, validated by time-dependent density functional theory calculations,
reveals that the signal is generated by the interference between multiple quantum pathways resonant with the excited state manifold.
The relative phase of such components is experimentally tuned by varying the probe chirp and we demonstrate how to decode the nuclear displacements along the different normal
modes from the experimentally detected impulsive Raman excitation profiles, thus revealing the multidimensional potential energy surfaces.
Accessing Excited State Molecular Vibrations by two-pulse Femtosecond Stimulated Raman Spectroscopy.
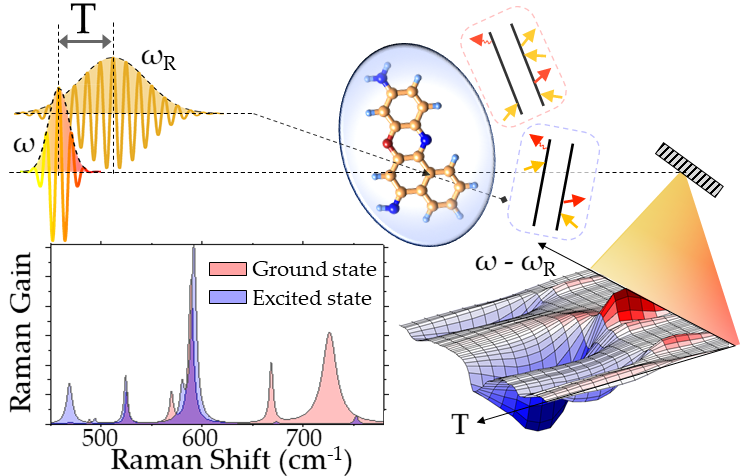
Assigning the measured vibrations to the pertaining ground or excited electronic states represents a demanding task for interpreting vibrational spectra recorded by time-resolved spectroscopic experiments.
Stimulated Raman scattering can coherently stimulate and probe molecular vibrations with optical pulses,
but it is generally restricted for the study of ground state properties. Two-pulse Femtosecond Stimulated Resonant Raman Scattering (FSRRS)
can be exploited for mapping excited state molecular properties: tuning the optical pulses to be in resonance with an electronic transition enables promoting the system to a targeted electronic state,
creating both excited state populations as well as excited state vibrational coherences.
Combining an experimental setup, which takes advantage of the the relative time delay between Raman and probe pulses for controlling the excited state contributions,
with a diagrammatic formalism able to dissect the concurring pathways that generate the Raman signal,
ground and excited state properties of the system under investigation can be simultaneously reconstructed.
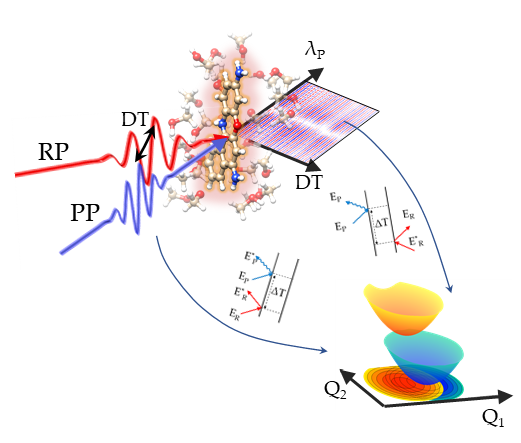
Mapping the interactions between vibrational and electronic molecular excitations by 2D Raman spectroscopy.
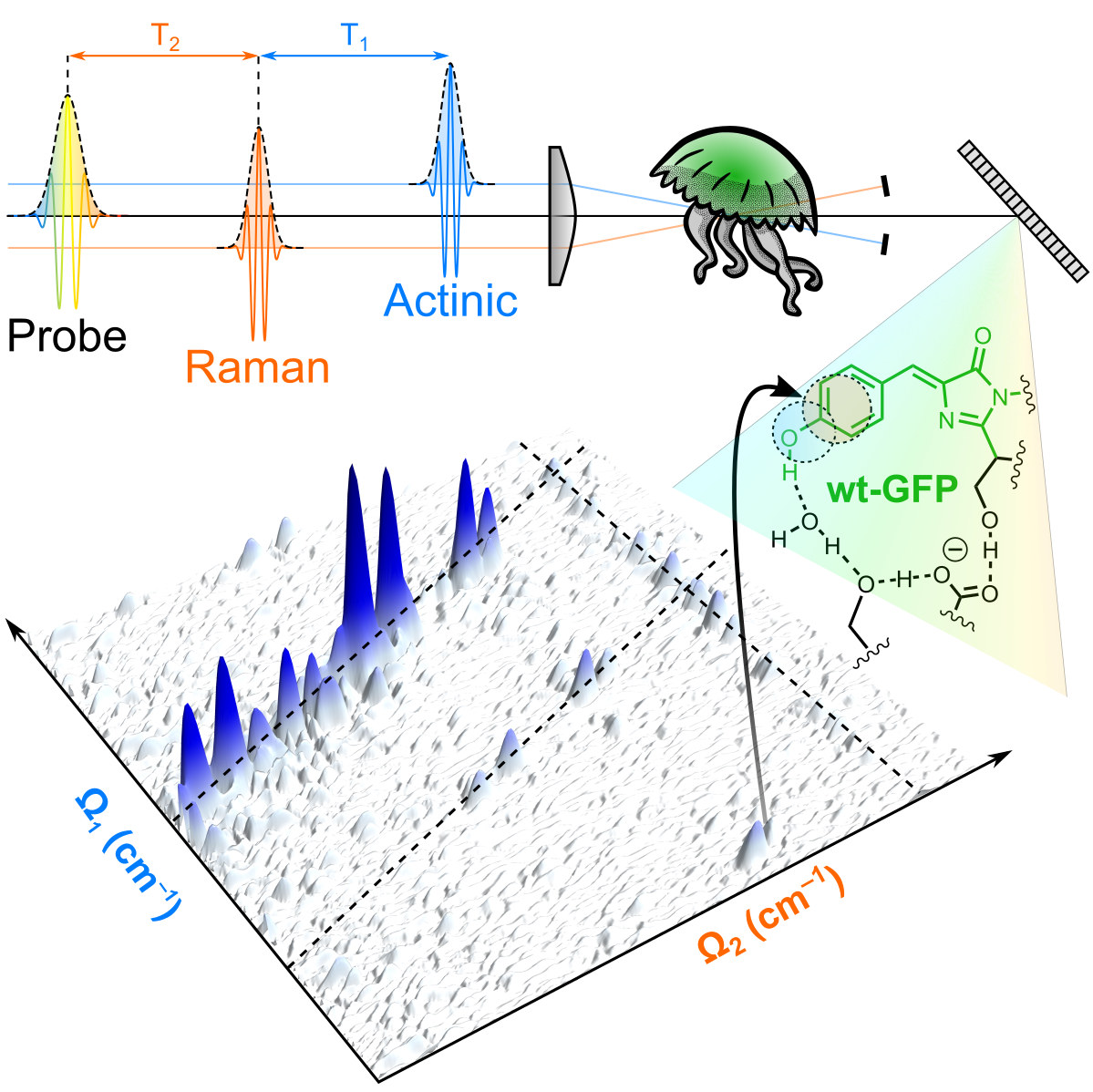
Light-induced processes in molecules rely on the efficient and directed conversion of photon energy into electronic and atomic motions.
This conversion is controlled by the underlying multidimensional energy surfaces,
which describe how the potential energy of the system changes with modifications in the vibrational and electronic configurations.
Mapping the potentianl energy surfaces over multiple vibrational dimensions discloses the ultrafast evolution of the system but is typically hampered by the need of spectroscopic probes
detecting different energy scales with high temporal and frequency resolution.
Two-Dimensional (2D) coherent Raman spectroscopy, by exploiting three temporally delayed femtosecond pulses to coherently generate and track excited-state vibrational wavepackets,
is able to probe vibrational correlations pertaining to a targeted electronically excited state.
The evolution of the wavepackets is determined by the shapes of the vibrationally structured potential energy surfaces of the system.
Couplings and correlations appear as cross peaks in 2D Raman maps, whose origin can be assigned by using a diagrammatic perturbative expansion in terms of the field-matter interactions,
mapping the multidimensional energy surfaces.
Atomic scale Truman show: making molecular movies of reacting proteins.
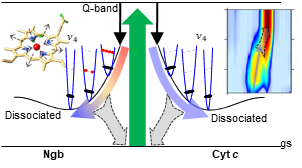
Identifying the structural rearrangement and the active sites in photo-induced reactions is a fundamental challenge to understand from a microscopic perspective the ultrafast dynamics of heme proteins.
Among the different types of hemes, those able to form the iron atom sixth bond with an internal residue, namely six-coordinate hemes,
represent one of the most elusive systems for ultrafast spectroscopies.
As dissociation of the internal residue is followed by a rapid rebinding embedded with several picosecond and femtosecond concurring processes,
such as structural reconfiguration, energy redistribution and relaxation on intermediate excited states, the natural hindrance for their study revolves around the simultaneous need of two key ingredients:
high temporal and spectral resolutions, which are mutually compromised due to the Heisenberg principle.
By exploiting femtosecond stimulated Raman spectroscopy (FSRS) to follow the ultrafast evolution of different six-coordinate hemes with combined high spectral resolution and femtosecond time precision,
we have determined the whole spectrum of vibrational amplitudes with extremely high temporal resolution,
allowing us to animate the molecular normal modes and to visualize how the different structural and thermal relaxation processes syncretize on the coherent picture of the reaction pathway.
International School On Nonlinear Vibrational Spectro-microscopy.
An International sChool On Nonlinear vibrational Spectro-microscopy (ICONS) will be held at the Physics Department of "Sapienza" University of Rome, July 30th-August 1st 2020.
ICONS will be structured around comprehensive review talks from major world leading experts in complementary areas of nonlinear Raman spectroscopy both on theory, experiments and applications.
The aim of the school is to provide high level-expertise for young researchers, graduate students interested in nonlinear Raman spectroscopy,
applied to access dynamical and microscopic properties at the molecular and condensed matter levels.
Specifically, the school will be three days of classes,
with an introduction on the peculiarities of nonlinear Raman approaches with respect to linear Raman spectroscopy,
discussing the technical requirements and the main tools that are conventionally exploited in modern experimental laboratories to manipulate and control optical pulses.
The following two days will be focused on time-resolved and microscopic techniques, with principles and examples.
The program, supported by world leading experts in the field of Raman nonlinear photonics, aims to have a balance between theoretical and experimental advances.
Exploiting Long pulses to probe Short phenomena.
Impulsive stimulated Raman scattering (ISRS) is a powerful technique able to real time monitor vibrational oscillations in photo-excited systems:
by combining a pump and a probe pulse for stimulating and then probing Raman coherences,
ISRS can directly access atomic motions and molecular properties.
Critically, at odd with frequency-domain Raman approaches,
ISRS typically requires long acquisition times since the system response has to be measured scanning a sequence of time delays between pump and probe pulses.
Introducing a chirp in the probe pulse, we have introduced a novel experimental scheme for the realization of Chirped-based ISRS (CISRS),
for recording the time-domain Raman information without scanning the pump and probe delay: since different probe wavelengths interact with sample at different time delays,
the evolution of the stimulated vibrational coherences is encoded in the probe spectrum.
Negative photo of graphene flakes by Cohenrent Raman Spectroscopy.
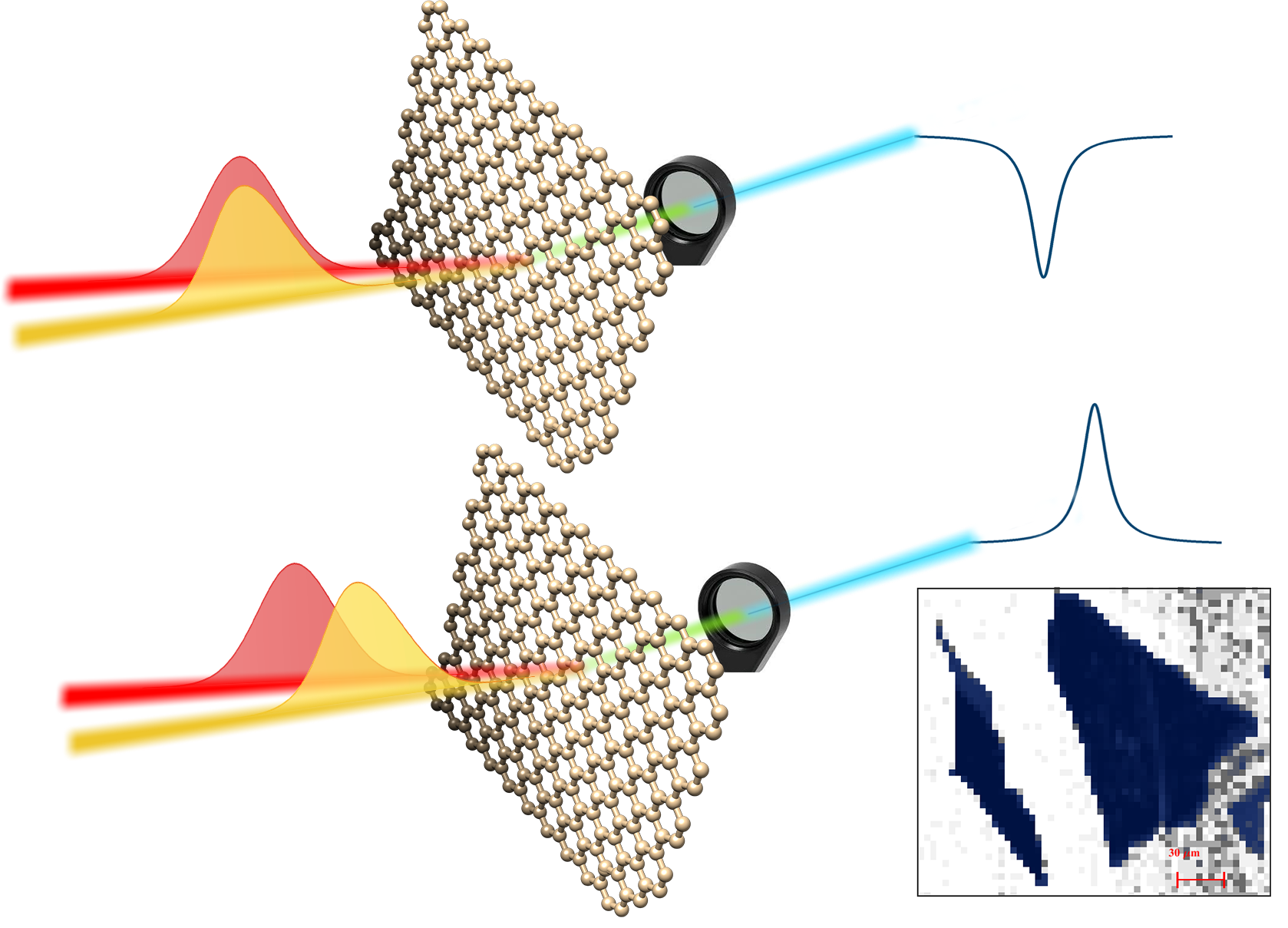
Spontaneous Raman spectroscopy is a powerful characterization tool for graphene research.
Its extension to the coherent regime, despite the large nonlinear third-order susceptibility of graphene, has so far proven challenging.
Due to its gapless nature, several interfering electronic and phononic transitions concur to generate its optical response, preventing to retrieve spectral profiles analogous to those of spontaneous Raman.
We have recently reported stimulated Raman spectroscopy of the G-phonon in single and multi-layer graphene, through coherent anti-Stokes Raman Scattering (CARS).
The nonlinear signal is dominated by a vibrationally non-resonant background, obscuring the Raman lineshape.
We demonstrate that the vibrationally resonant coherent anti-Stokes Raman Scattering peak can be measured by reducing the temporal overlap of the laser excitation pulses,
suppressing the vibrationally non-resonant background. Modelling the spectra, for taking into account the electronically resonant nature of both,
we have shown how CARS can be used for graphene imaging with vibrational sensitivity.
Manipulating magnetic properties with femtosecond laser pulses.
Raman techniques are pivotal in many interdisciplinary aspects of photonics.
In turn, fundamentals of non-linear photonics can also be used as a key enabling tools to disclose sub-100 fs magnetic excitations dynamics exploiting coherent Raman spectroscopy.
The relevance of probing the ultrafast dynamics of the exchange interaction strikes a chord of interest from both the fundamental and applied science standpoint.
On the one hand one wonders on how fast angular momentum can be exchanged, and if it may become possible to reach the timescale of the spin-orbit interaction.
On the other hand, the study of the fundamental and practical limits of the speed of manipulation of the magnetic ordering is obviously of great importance for magnetic recording
and information processing technologies.
Articles
2013
2014
2015
2016
2017
2018
2019
2020
2021
2022
2023
2024
2025
2026
Published
Accepted
Submitted
2026
Vibrational Dynamics à la Carte: Background-Free Mode-Selectivity by Phase-Controlled Impulsive Raman Spectroscopy.
G. Batignani, A. Mariani, M. Martinati, E. Mai, M. M. Neethish and T. Scopigno.
Submitted, , (2026).
2025
Perylene bisimide derivatives in water solutions, effects of salt on aggregation kinetics and spectroscopic properties.
M. Martinati, G. Batignani, G. De Maria, G. Fabrizi, Y. Gazzilli, V. Pennacchietti, F. Yang, A. Boffi, T. Scopigno and R. Piacentini.
Submitted, , (2025).
Real-Time Tracking of the intramolecular vibrational dynamics of liquid water.
G. Giovannetti, S. Ryabchuk, A. Bin Wahid, H.-Y. Chen, G. Batignani, E. P. Månsson, O. Cannelli, E. Mai, A. Trabattoni, O. Neufeld, A. Rubio, V. Wanie,
H. Marroux, T. Scopigno, M. Chergui, and F. Calegari.
Communications Chemistry, , (2025).
Epsilon-near-zero nonlinearity enhancement in the extreme ultraviolet.
C. Ferrante, E. Principi, L. Assogna, A. Sahoo, G. Batignani, G. Fumero, L. Foglia, R. Mincigrucci, L. Giannessi, T. Scopigno, C. Masciovecchio, and A. Marini.
Light: Science & Applications, 14, (2025).
Solving ZIB Challenges: The Dynamic Role of Water in Deep Eutectic Solvents electrolyte.
E. Emanuele, G. Batignani, G. Cerullo G. Leita, E. Mai, M. Martinati, C. Mele, M. M. Neethish, T. Scopigno, B. Bozzini.
Journal of Materials Chemistry A, 13, 9778-9790 (2025).
2024
Orchestrating nuclear dynamics in a permanganate doped crystal with Chirped Pump-Probe spectroscopy.
E. Mai, P. Malakar, G. Batignani, M. Martinati, S. Ruhman and T. Scopigno.
The Journal of Physical Chemistry Letters, 15 (2024).
Femtosecond Stimulated Raman Spectroscopy.
G. Batignani, C. Ferrante, G. Fumero, M. Martinati and T. Scopigno.
Nature Reviews Methods Primers, 4, 34 (2024).
2023
Temperature dependence of coherent versus spontaneous Raman scattering.
G. Batignani, E. Mai, M. Martinati, S. Mukamel and T. Scopigno.
Physical Review Letters 133, 206902 (2024).
Retrieving genuine nonlinear Raman responses in ultrafast spectroscopy via deep learning.
G. Fumero, G. Batignani, E. Cassetta, C. Ferrante, S. Giagu and T. Scopigno.
APL Photonics, 9, 066119 (2024).
2022
Absolute excited state molecular geometries revealed by resonance Raman signals.
G. Batignani, E. Mai, G. Fumero, S. Mukamel and T. Scopigno.
Nature Communications, 13, 7770 (2022)
2-hydroxyisobutyrate (2-HIBA), an unspecified mammalian metabolite, modulates ageing and fat deposition depending on the high glucose diet in C. elegans animal model.
E. Schifano, G. Conta, A. Preziosi, C. Ferrante, G. Batignani, A. Tomassini, F. Sciubba, T. Scopigno, D. Uccelletti and A. Miccheli.
Frontiers in Molecular Biosciences, 9, (2022)
Picosecond energy transfer in a transition metal dichalcogenide-graphene heterostructure revealed by transient Raman spectroscopy.
C. Ferrante, G. Di Battista, L. E. Parra Lopez, G. Batignani,
E. Lorchat, A. Virga, S. Berciaud and T. Scopigno.
PNAS, 119, 15, e2119726119, (2022)
Stimulated Raman lineshapes in the large light-matter interaction limit.
G. Batignani, G. Fumero, E. Mai, M. Martinati and T. Scopigno.
Optical material X, 13, 100134, (2022)
2021
Excited-state energy surfaces in molecules revealed by impulsive stimulated Raman excitation profiles.
G. Batignani, C. Sansone, C. Ferrante, G. Fumero, S. Mukamel and T. Scopigno.
The Journal of Physical Chemistry Letters, 12, 9239–9247, (2021)
Non-linear self-driven spectral tuning of Extreme Ultraviolet Femtosecond Pulses in monoatomic materials.
C. Ferrante, E. Principi, A. Marini, G. Batignani et al.
Light: Science & Applications, 10, 92, (2022)
2020
Accessing Excited State Molecular Vibrations by Femtosecond Stimulated Raman Spectroscopy.
G. Batignani, C. Ferrante and T. Scopigno.
The Journal of Physical Chemistry Letters, 11, 18, 7805, (2020)
Ultrafast dynamics and vibrational relaxation in six-coordinate heme proteins revealed by Femtosecond Stimulated Raman Spectroscopy.
C. Ferrante and G. Batignani and E. Pontecorvo, L.C. Montemiglio, M. H. Vos and T. Scopigno.
Journal of the American Chemical Society, 142, 2285, (2020)
Two-dimensional impulsively stimulated resonant Raman spectroscopy of molecular excited-states.
G. Fumero, C. Schnedermann, G. Batignani, T. Wende, M. Liebel, G. Bassolino, C. Ferrante, S. Mukamel, P. Kukura, T. Scopigno.
Physical Review X, 10, 011051, (2020)
2019
Broadband Impulsive Stimulated Raman Scattering based on a Chirped Detection.
G. Batignani, C. Ferrante, G. Fumero and T. Scopigno.
The Journal of Physical Chemistry Letters, 10, 7789, (2019)
Modeling the ultrafast response of two-magnon Raman excitations in antiferromagnets on the femtosecond timescale.
G. Batignani, E. Pontecorvo, D. Bossini, C. Ferrante, G. Fumero, G. Cerullo, S. Mukamel and T. Scopigno.
Annalen der Physik, 1900439, (2019)
Coherent anti-Stokes Raman Spectroscopy of single and multi-layer graphene.
A. Virga, C. Ferrante, G. Batignani, D. De Fazio, A. D. Nunn, A. C. Ferrari, G. Cerullo, T. Scopigno.
Nature Communications, 10, 3658, (2019)
Genuine dynamics vs cross phase modulation artefacts in Femtosecond Stimulated Raman Spectroscopy.
G. Batignani, G. Fumero, E. Pontecorvo, C. Ferrante, S. Mukamel, T. Scopigno.
ACS Photonics, 6, 492, (2019)
The Potential of EuPRAXIA@SPARC_LAB for Radiation Based Techniques.
A. Balerna, S. Bartocci, G. Batignani et al.
Condensed Matter, 4, 30, (2019)
2018
Probing Femtosecond Lattice Displacement upon Photo-carrier generation in Lead Halide Perovskite.
G. Batignani, G. Fumero, A. R. S. Kandada, G. Cerullo, M. Gandini, C. Ferrante, A. Petrozza, T. Scopigno.
Nature Communications, 9, 1971, (2018)
Resonant Broadband Stimulated Raman scattering in Myoglobin.
C. Ferrante, G. Batignani, G. Fumero, E. Pontecorvo, A. Virga, L. C. Montemiglio, G. Cerullo, M. H. Vos, T. Scopigno.
Journal of Raman Spectroscopy, 1-8, (2018)
2017
Manipulating impulsive stimulated Raman spectroscopy with a chirped probe pulse.
L. Monacelli, G. Batignani, G. Fumero, C. Ferrante, S. Mukamel and T. Scopigno.
The Journal of Physical Chemistry Letters, 8, 966, (2017)
2016
Visualizing excited-state dynamics of a diaryl thiophene: femtosecond stimulated Raman scattering as a probe of conjugated molecules.
G. Batignani, E. Pontecorvo, C. Ferrante, M. Aschi, C.G. Elles and T. Scopigno.
The Journal of Physical Chemistry Letters, 7, 2981, (2016)
Probing ultrafast processes by fifth order Stimulated Raman Scattering.
G. Fumero, G. Batignani, K. E. Dorfman, S. Mukamel and T. Scopigno.
Journal of Physics: Conference Series, 689, 012023, (2016)
Probing ultrafast photo-induced dynamics of the exchange energy in a Heisenberg antiferromagnet.
G. Batignani, D. Bossini, N. Di Palo, C. Ferrante, E. Pontecorvo, G. Cerullo, A. Kimel and T. Scopigno.
OSA Technical Digest, http://dx.doi.org/10.1364/UP-2016-UTh3A.7, (2016)
Electronic resonances in broadband stimulated Raman spectroscopy.
G. Batignani, E. Pontecorvo, G. Giovannetti, C. Ferrante, G. Fumero and T. Scopigno.
Scientific Reports, 6, 18445, (2016)
2015
On the resolution limit of Femtosecond Stimulated Raman Spectroscopy: modelling fifth-order signals with overlapping pulses.
G. Fumero, G. Batignani, K. E. Dorfman, S. Mukamel and T. Scopigno.
Chem. Phys. Chem., 16, 3438, (2015)
Probing ultrafast photoinduced dynamics of the exchange energy in an Heisenberg antiferromagnet.
G. Batignani, D. Bossini, N. Di Palo, C. Ferrante, E. Pontecorvo, G. Cerullo, A. Kimel and T. Scopigno.
Nature Photonics, 9, 506, (2015)
Energy flow between spectral components in 2D Broadband Stimulated Raman Spectroscopy.
G. Batignani, G. Fumero, S. Mukamel and T. Scopigno.
Physical Chemistry Chemical Physics, 17, 10454, (2015)
2014
Snapshots of sub-ps dynamics in heme-proteins captured by Femtosecond Stimulated Raman Scattering.
T. Scopigno, C. Ferrante, E. Potecorvo, G. Batignani.
OSA Technical Digest, http://dx.doi.org/10.1364/UP.2014.11.Fri.A.6, (2014)
2013
Detection of excited level population transfer in an MOT through the measurement of trapped atom number.
L. Moi, G. Batignani, A. Khanbekyan et al.
Measurement Science and Technology , 24, 015201, (2013)
Teaching
Academic Year 2025/2026
CORSO DI LAUREA TRIENNALE: “FISICA 1”
Laurea Triennale in Scienze Chimiche
Dipartimento di Chimica, Sapienza Università di Roma
Pagina Web del Corso.
CORSO DI LAUREA TRIENNALE: “LABORATORIO DI MECCANICA 1”
Laurea Triennale in Fisica
Dipartimento di Fisica, Sapienza Università di Roma
Pagina Web del Corso.
Academic Year 2024/2025
CORSO DI LAUREA TRIENNALE: “FISICA 1”
Laurea Triennale in Scienze Chimiche
Dipartimento di Chimica, Sapienza Università di Roma
Pagina Web del Corso.
Academic Year 2022/2023
CORSO DI LAUREA TRIENNALE: “LABORATORIO DI CALCOLO”
Laurea Triennale in Fisica
Dipartimento di Fisica, Sapienza Università di Roma
Pagina Web del Corso.
Academic Year 2021/2022
CORSO DI LAUREA TRIENNALE: “LABORATORIO DI CALCOLO”
Laurea Triennale in Fisica
Dipartimento di Fisica, Sapienza Università di Roma
Pagina Web del Corso.
Academic Year 2020/2021
CORSO DI LAUREA TRIENNALE: “OTTICA E LABORATORIO”
Laurea Triennale in Fisica
Dipartimento di Fisica, Sapienza Università di Roma
Pagina Web del Corso.
Academic Year 2019/2020
CORSO DI LAUREA TRIENNALE: “OTTICA E LABORATORIO”
Laurea Triennale in Fisica
Dipartimento di Fisica, Sapienza Università di Roma
Pagina Web del Corso.
Percorsi di eccellenza
PROPOSTA DI APPROFONDIMENTO TEORICO/SPERIMENTALE:
Verso il limite del singolo ciclo ottico, sintesi coerente di impulsi laser ultrabrevi di 10 fs.
Il percorso prevede una prima parte dove verranno introdotti i metodi teorici/computazionali da impiegare per simulare numericamente
la propagazione di fasci laser.
Seguirà uno stage in laboratorio, dove partendo da una sorgente laser pulsata, verranno sintetizzati impulsi tunabili in lunghezza d’onda, che verranno compressi temporalmente sino ad arrivare a durate prossime al singolo ciclo ottico.
Gli impulsi ottenuti verranno caratterizzati tramite misure di autocorrelazione, che potranno essere validate ed analizzate tramite i protocolli computazionali studiati nella prima parte.
Thesis
MASTER THESIS
Presso i Laboratory Femtoscopy sono disponibili dissertazioni magistrali su:
DISSERTAZIONI
Sono disponibili dissertazioni triennali sui seguenti argomenti:
- Beam Propagation Method applicato alla propagazione di luce laser.
- Propagazione di impulsi di luce laser tramite l'Equazione di Schrödinger non lineare.
- Approccio Diagrammatico allo scattering Raman.
- Spettroscopia Ultraveloce Tramite Assorbimento Transiente.
- Spettroscopia Raman Risolta in Tempo.
- Effetto Raman Stimolato mediante impulsi laser al Femtosecondo.
- Laser Cooling e Intrappolamento Magneto-Ottico.
- Diffrazione della luce in regime di Fresnel.
- Spetroscopia Multi-Dimensionale.
- Effetto Raman Coerente per Esperimenti di Microscopia.
- Raman Time-Domain vs Frequency-Domain.
Per maggiori informazioni contattare il docente.
Dissertazioni Discusse
- "
Stabilizzazione di perovskiti fotovoltaiche attraverso intercalazione di gas nobili ad alta pressione",
Eleonora Polini - A.A. 16/17 (Seminario).
- "
Metodo Hartree-Fock molecolare: implementazione Matlab per molecole biatomiche a due elettroni",
Giovanni Caldarelli - A.A. 17/18.
- "
Raffreddamento atomico e condensati Bose-Einstein",
Amer Omar - A.A. 18/19.
- "
Laser cooling di atomi neutri",
Nicola Fiorente - A.A. 18/19.
- "
Doppler cooling e trappole magneto-ottiche per atomi neutri",
Francesco Macchioni - A.A. 18/19.
- "
Spettroscopia vibrazionale impulsiva nel dominio del tempo",
Giorgio Minati - A.A. 19/20.
- "
Studio dello scattering Raman spontaneo e stimolato nel formalismo di seconda quantizzazione",
Irene Gaiardoni - A.A. 19/20.
- "
Spatially offset Raman spectroscopy, principi e applicazioni",
Fabrizio Fatelli - A.A. 19/20.
- "
Laser cooling e intrappolamento magneto-ottico di atomi e molecole biatomiche",
Alessandro Petrini - A.A. 19/20.
- "Molecular potential energy surfaces by impulsive stimulated Raman excitation profiles",
Carlotta Sansone - A.A. 20/21. Article.
- "Effetto Raman nel formalismo della seconda quantizzazione",
Nicolò Prosperi - A.A. 20/21.
- "Measuring the phase of Raman susceptibilities by Impulsive Vibrational Spectroscopy reveals excited-state displacements",
Emanuele Mai - A.A. 20/21. Article.
- "Dipendenza del segnale di assorbimento e scattering Raman in funzione del displacement molecolare",
Dalila Di Serio - A.A. 20/21.
- "Laser Cooling e Trappole Magneto Ottiche",
Fabrizio Spera - A.A. 21/22.
- "Spettroscopia Raman Risonante e dipendenza dai displacement molecolari",
Francesco Costantini - A.A. 21/22.
- "Intrappolamento magneto-ottico e condensazione di Bose-Einstein",
Luca Pancione - A.A. 21/22.
- "Spettroscopia Raman impulsiva nel dominio temporale",
Pietro Todesco - A.A. 21/22.
- "Dispersione cromatica e propagazione di impulsi laser ultracorti",
Francesco Vergari - A.A. 21/22.
- "Raman Induced Kerr Effect: a study on the polarization dependent non linear vibrational responses",
Paolo Fachin - A.A. 21/22. Article.
- "Spettroscopia Raman nel dominio delle frequenze ed in trasformata di Fourier",
Matteo Cacioppo - A.A. 21/22.
- "Beam Propagation Method applicato alla propagazione della luce",
Giovanni Presutti - A.A. 22/23.
- "Effetti non lineari nella propagazione di impulsi di luce",
Lorenzo Fiordaliso - A.A. 22/23.
- "Excited state dynamics of Fucoxanthin studied by femtosecond stimulated Raman scattering",
Alfredo Tarantino - A.A. 22/23.
- "Metodo di Hartree-Fock applicato al trimero omonucleare",
Carlo Sabatini - A.A. 22/23.
- "Approccio numerico alla propagazione della luce: Beam Propagation Method",
Pietro Todesco - A.A. 22/23.
- "Effetto Raman impulsivo nel dominio del tempo: Principi e Applicazioni",
Filippo Maccarone - A.A. 22/23.
- "Beam propagation method applicato alla propagazione della luce in mezzi turbolenti",
Matteo Spaziani - A.A. 22/23.
- "Spettroscopia Raman stimolata al femtosecondo: principi e applicazioni",
Luigi Graziano - A.A. 22/23.
- "Propagazione di impulsi laser ultracorti in materiali dispersivi",
Viola Valeau - A.A. 22/23.
- "Unravelling excited-state molecular vibrations by chirped Raman spectroscopy",
Assia Mariani - A.A. 22/23. Article.
- "Impulsive Stimulated Raman Spectroscopy at 50 kHz repetition rate",
Florian Pagnini - A.A. 23/24.
- "Studio numerico della propagazione della luce attraverso il Beam Propagation Method",
Virginia Protano - A.A. 23/24.
- "Artificial Intelligence-driven data analysis for nonlinear vibrational spectroscopy",
Pietro Lussana - A.A. 24/25. Article.
- "Applicazioni di Femtochimica",
Siria Domeniconi - A.A. 24/25.
- "Temperature Dependence of Stimulated Resonant Raman Scattering: Thoery and Experiment",
Gaia De Maria - A.A. 24/25.
- "Generating Spectrally Tunable near-UV Femtosecond Pulses for Pump-Probe Spectroscopies",
Lorenzo Perot - A.A. 24/25.
- "Exploring singlet to triplet ultrafast dynamics in Thiouracil via pump-probe vibrational spectroscopy",
Irene Rea - A.A. 24/25.
- "Effetti del confinamento spazio-temporale nella propagazione di impulsi luminosi",
Luna Tarola - A.A. 24/25.
- "Effetto Raman: il ruolo delle risonanze elettroniche",
Edoardo Zaccaria - A.A. 24/25.
Contacts
Mail: giovanni.hiddenbatignani[at]hiddenuniroma1.it
Address: Piazzale Aldo Moro 5, Roma, 00185, Italy
Office: Edificio "E. Fermi", Room 508
Office hours: any time (if I am free).
Phone (office): 0649913478
Phone (lab):




.png)